隨著現(xiàn)代科技的飛速發(fā)展,新型電子產(chǎn)品以及新能源汽車的出現(xiàn),人們對(duì)鋰電池的容量,能量密度以及安全性提出了更高標(biāo)準(zhǔn)的要求。鋰電池安全高效的運(yùn)行離不開(kāi)電解質(zhì)的穩(wěn)定運(yùn)作,其基本功能是在電池的正負(fù)極之間作為內(nèi)部電荷傳遞的主要介質(zhì),并防止電池發(fā)生內(nèi)短路現(xiàn)象。理想的電解質(zhì)應(yīng)該促進(jìn)電極表面的法拉第反應(yīng)動(dòng)力學(xué)過(guò)程,同時(shí)緩解鋰電池界面處的副反應(yīng)。傳統(tǒng)的碳酸酯基電解液體系由于其較高的脫溶劑化能壘和較差的界面相容性,通常會(huì)導(dǎo)致電極界面緩慢的反應(yīng)動(dòng)力學(xué)和較大的副反應(yīng)。
最近,浙江大學(xué)材料學(xué)院陳立新教授、范修林研究員團(tuán)隊(duì)提出了一種新的分子對(duì)接電解液(MDE)設(shè)計(jì)策略,該策略克服了鋰鹽在隱性溶劑分子(具有潛在的離子配位官能團(tuán),但是由于空間位阻等導(dǎo)致無(wú)法溶劑化鋰離子)中解離的局限性,同時(shí)在電極界面處實(shí)現(xiàn)了快速穩(wěn)定的電化學(xué)反應(yīng)。
該項(xiàng)研究成果于北京時(shí)間2024年7月15日,發(fā)表在國(guó)際頂級(jí)期刊《Nature Chemistry》。論文第一作者為浙江大學(xué)馬寶琛和張海闊博士研究生,通訊作者為浙江大學(xué)范修林研究員,并受到騰訊優(yōu)圖實(shí)驗(yàn)室,浙江大學(xué)陳立新教授、肖學(xué)章副教授和馬里蘭大學(xué)鄧濤博士(現(xiàn)為上海交大中英低碳研究院副教授)的大力支持。浙江大學(xué)為該論文的唯一通訊單位。
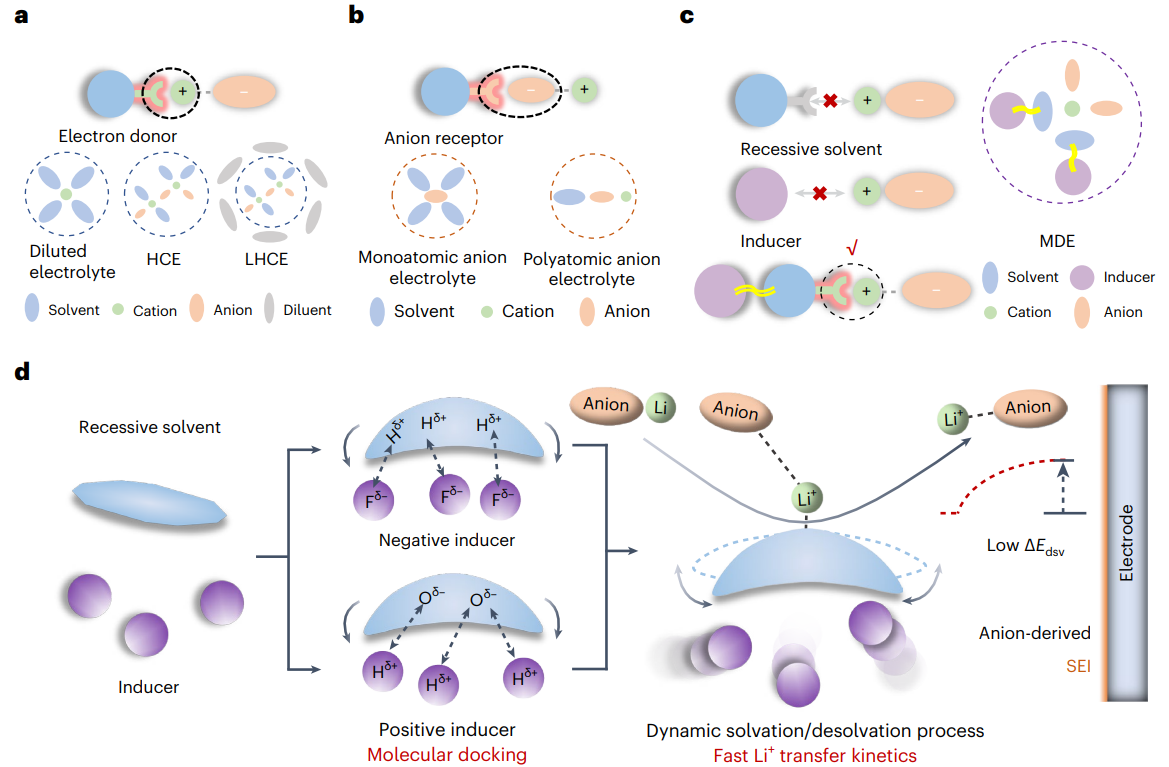
圖1 目前的電解液設(shè)計(jì)所用的顯性溶劑化(a,b)與本文提出的隱性溶劑化模型(c,d)示意圖
傳統(tǒng)電解液體系目前主要基于兩種機(jī)制:電子供體或陰離子受體。根據(jù)其特定的溶劑化結(jié)構(gòu),可進(jìn)一步分為稀電解液、高濃電解液、局部高濃電解液、單原子陰離子電解液以及多原子陰離子電解液等。以上電解液體系都是通過(guò)自發(fā)的離子-溶劑配位形成的,稱為顯性溶劑化。然而,在隱性溶劑化模型中,單獨(dú)的隱性溶劑或誘導(dǎo)劑不能溶解鋰鹽,自然也不具備離子電導(dǎo)率支撐電池的正常運(yùn)行。當(dāng)隱性溶劑與某些特定的氟苯類或鹵代烷類化合物(稱為誘導(dǎo)劑)混合時(shí),誘導(dǎo)劑分子會(huì)通過(guò)非典型氫鍵相互作用(特別是Fδ--Hδ+或Hδ+-Oδ-相互作用)使得隱性溶劑分子產(chǎn)生構(gòu)型變化,影響分子表面靜電勢(shì)能分布,進(jìn)而激活隱性溶劑分子的Li+配位點(diǎn)。這些分子間的動(dòng)態(tài)相互作用形成了Li+-溶劑間的動(dòng)態(tài)配位過(guò)程,降低了Li+界面處的脫溶劑化能壘,同時(shí)提高了陰離子界面還原動(dòng)力學(xué),緩解了電解液溶劑分子在正/負(fù)極側(cè)的副反應(yīng)。同時(shí),誘導(dǎo)劑調(diào)控的離子動(dòng)態(tài)脫溶劑化過(guò)程也使得Li+-FSI+離子對(duì)配位能力增強(qiáng),從而在電極側(cè)形成陰離子衍生的SEI。
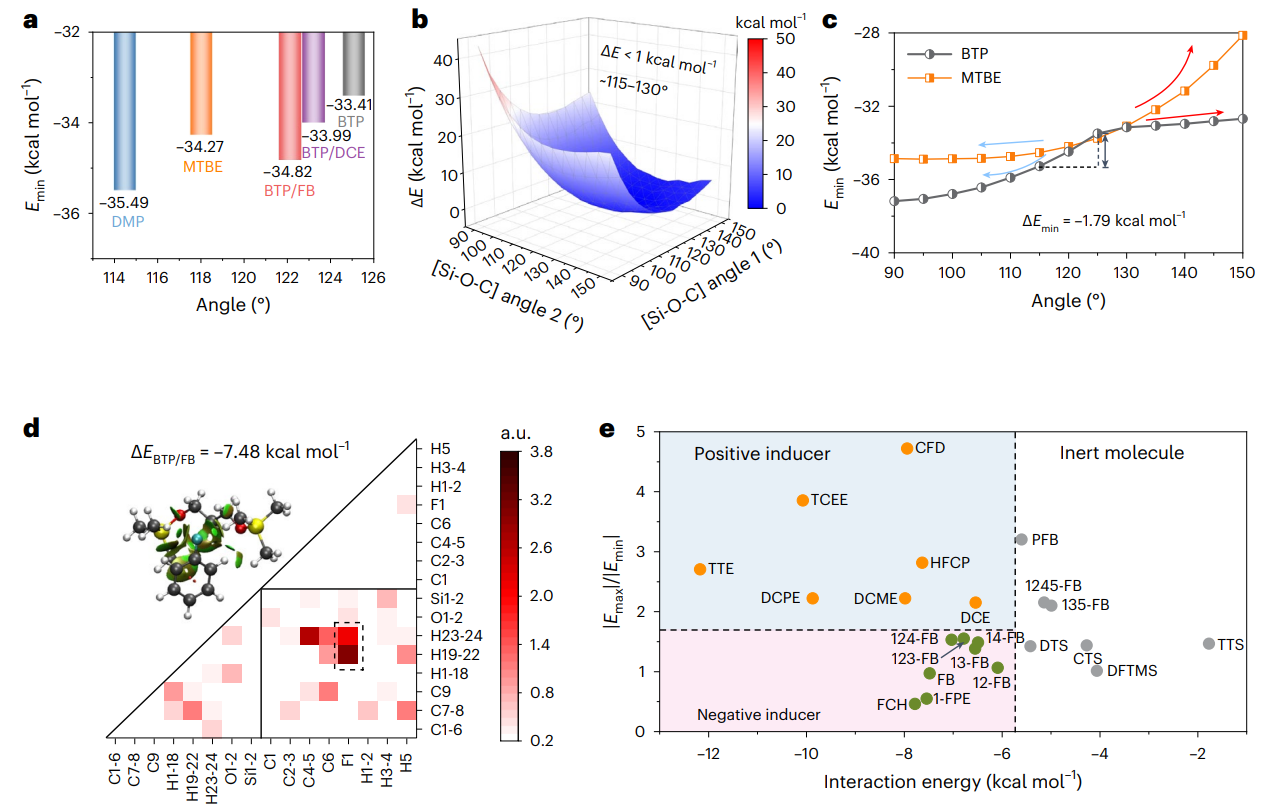
圖2 隱性溶劑與誘導(dǎo)劑相關(guān)計(jì)算模擬
為了全面的解析分子對(duì)接機(jī)制和結(jié)構(gòu)-性能的對(duì)應(yīng)關(guān)系,選擇1,3-雙(三甲基硅氧基)丙烷(BTP)以及1,2-雙(三甲基硅氧基)乙烷(BTE)作為典型的隱性溶劑分子,基于自然鍵軌道以及分子靜電勢(shì)能分析,揭示了隱性溶劑分子構(gòu)型、空間位阻、鍵角大小等與離子配位能力的內(nèi)在關(guān)系,提出誘導(dǎo)劑作用力閾值(< 5.7 kcal/mol)并結(jié)合實(shí)驗(yàn)結(jié)果,篩選并設(shè)計(jì)出了15種氟苯、鹵代烷烴類誘導(dǎo)劑分子。結(jié)合核磁、紅外光譜、拉曼、等溫量熱滴定法、飛秒瞬態(tài)吸收光譜等測(cè)試表征手段進(jìn)一步證明了隱性溶劑分子與誘導(dǎo)劑之間的分子對(duì)接機(jī)制。
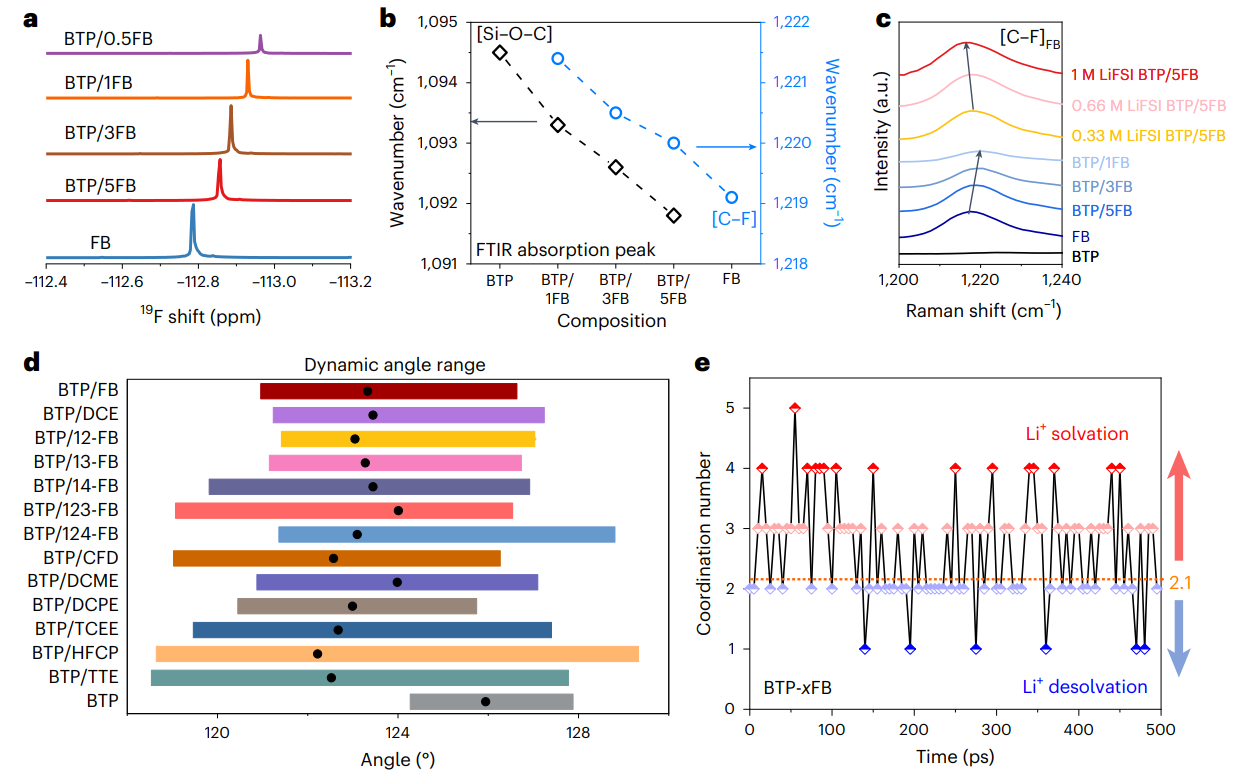
圖3 分子對(duì)接電解液測(cè)試表征與模擬計(jì)算
通過(guò)密度泛函理論(DFT)與分子動(dòng)力學(xué)(MD)模擬發(fā)現(xiàn),相較于純隱性溶劑BTP分子,誘導(dǎo)劑的引入會(huì)使得隱性溶劑分子[Si-O-C]鍵角向小角度壓縮,同時(shí)分子中O位點(diǎn)靜電勢(shì)能減小,對(duì)鋰離子的配位能力增強(qiáng)。與此同時(shí),由于誘導(dǎo)劑分子在隱性溶劑分子周圍的熱運(yùn)動(dòng),誘導(dǎo)劑的配位數(shù)會(huì)出現(xiàn)明顯的隨機(jī)波動(dòng),高配位數(shù)(≥3)的誘導(dǎo)劑有利于Li+的溶劑化,低配位數(shù)(1或2)的誘導(dǎo)劑有利于Li+自發(fā)脫離溶劑化殼結(jié)構(gòu)。因此,MDE通過(guò)誘導(dǎo)劑與隱性溶劑分子間的動(dòng)態(tài)對(duì)接/解耦過(guò)程,導(dǎo)致Li+-溶劑相互作用的動(dòng)態(tài)溶劑化/脫溶劑化過(guò)程,有助于降低Li+脫溶劑化能壘,實(shí)現(xiàn)快速的Li+界面電化學(xué)反應(yīng)。
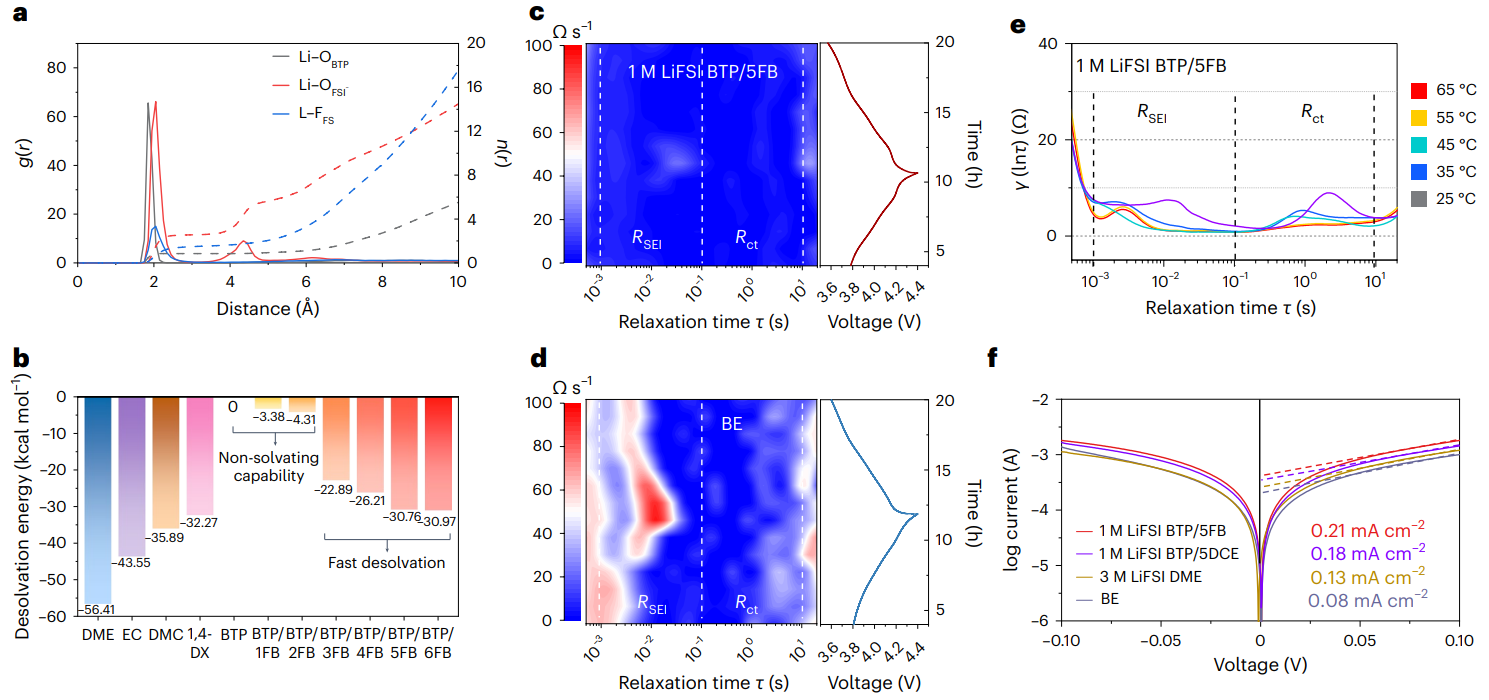
圖4 動(dòng)態(tài)鋰離子溶劑配位實(shí)現(xiàn)快速的傳輸動(dòng)力學(xué)
進(jìn)一步的DFT、MD計(jì)算模擬以及弛豫時(shí)間分析實(shí)驗(yàn)證明了相較于傳統(tǒng)的酯基電解液,MDE中較低的離子脫溶劑化能壘以及生成低界面阻抗的SEI,有利于提高MDE對(duì)于鋰金屬界面的兼容性。
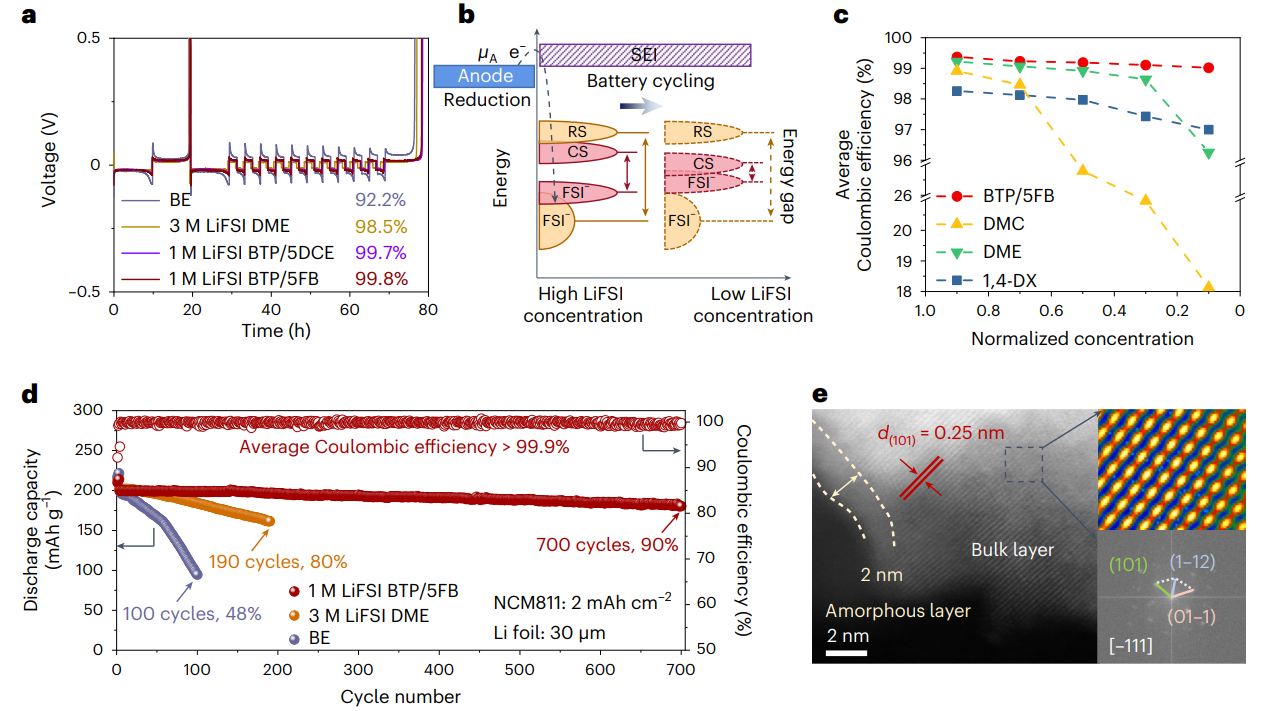
圖5 鋰金屬負(fù)極以及全電池循環(huán)性能分析
基于分子對(duì)接電解液的設(shè)計(jì)策略,電解液的鋰銅庫(kù)倫效率可超過(guò)99.5%,在4.4 V 30 μm Li||2.0 mAh cm-2 LiNi0.8Co0.1Mn0.1O2(NCM811)全電池中,700次循環(huán)后的容量保持率為90%。在1 Ah 3.4 mAh cm-2石墨||3.0 mAh cm-2 NCM811軟包電池中,550次循環(huán)后的容量保持率為98%。同時(shí)該策略設(shè)計(jì)的電解液對(duì)鋰離子電池體系也具有良好的適配性。
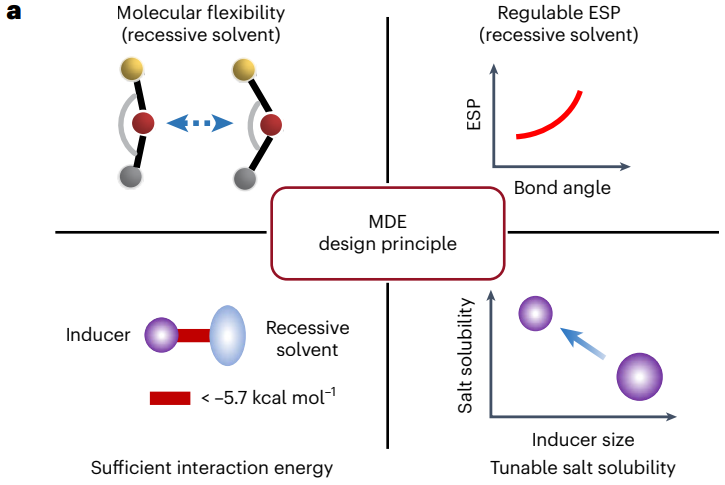
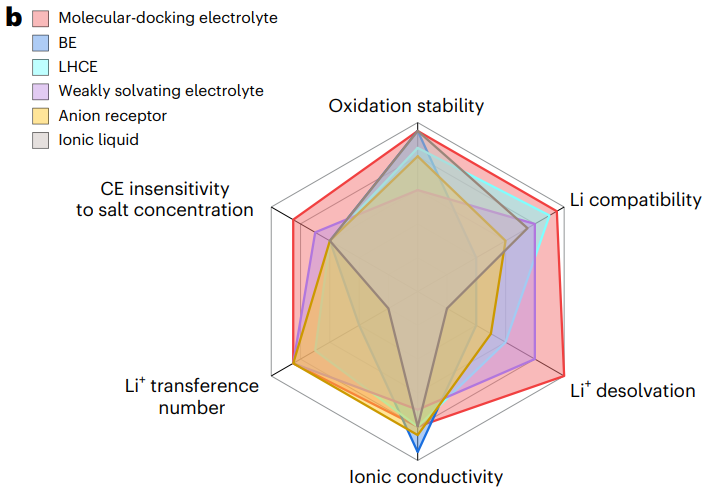
圖6 分子對(duì)接電解液的設(shè)計(jì)原則和相關(guān)指標(biāo)評(píng)價(jià)
在這項(xiàng)工作中,作者提出了一種基于分子對(duì)接機(jī)制的新型電解液設(shè)計(jì)策略,有望提升鋰離子/金屬電池中界面動(dòng)力學(xué)和同時(shí)抑制界面副反應(yīng),實(shí)現(xiàn)鋰電池的高效穩(wěn)定循環(huán)。設(shè)計(jì)MDE的關(guān)鍵因素包括隱性溶劑的獨(dú)特分子柔性(例如,BTP硅氧烷分子的絕熱彎曲勢(shì)能< 1.7 kcal/mol),同時(shí)分子鍵角的變化對(duì)應(yīng)著分子靜電勢(shì)能的調(diào)節(jié)(例如在分子鍵角減小時(shí)溶劑配位官能團(tuán)的靜電勢(shì)較低),以及非典型氫鍵(Fδ--Hδ+或Hδ+-Oδ-相互作用)引起的足夠的分子間相互作用(< -5.7 kcal/mol)。此外,使用更小尺寸的誘導(dǎo)劑,減小分子對(duì)接過(guò)程中的位阻效應(yīng),可以進(jìn)一步提升鋰鹽在隱性溶劑中的溶解度。
